
Die Stadieneinteilung bei chronischer lymphatischer Leukämie ist kein reiner Formalismus. Sie hilft, den Verlauf einzuordnen, die Kontrollen sinnvoll zu planen und zu verstehen, warum viele Betroffene trotz Diagnose zunächst keine Therapie brauchen. In diesem Beitrag geht es um die CLL-Stadien, ihre Aussagekraft für Verlauf und Prognose sowie darum, welche Laborwerte und Symptome heute oft mehr zählen als die Zahl im Befund. Ich schreibe bewusst praxisnah, weil genau dort die meisten Missverständnisse entstehen.
Die Einordnung von CLL bestimmt den Fahrplan, aber nicht das Schicksal
- Binet und Rai sind die beiden klassischen klinischen Systeme, in Europa meist Binet.
- Für die Stadieneinteilung reichen in der Regel körperliche Untersuchung und Blutbild; CT oder MRT ändern das Stadium nicht.
- Ein frühes Stadium bedeutet oft Beobachtung statt sofortiger Therapie.
- Für die Prognose sind TP53, IGHV, β2-Mikroglobulin und der komplexe Karyotyp entscheidend.
- Lymphozytenzahl allein ist kein ausreichender Behandlungsgrund.
- Wer Symptome, Blutwerte und genetische Marker zusammen liest, bekommt ein deutlich realistischeres Bild als mit dem Stadium allein.
Wie die Stadieneinteilung bei CLL aufgebaut ist
Wenn ich über die CLL-Stadien spreche, meine ich zwei klinische Systeme, die sich seit Jahren bewährt haben: Binet und Rai. Beide beschreiben nicht einfach, wie viele Zellen im Blut zirkulieren, sondern vor allem, ob Lymphknoten, Milz, Leber und Knochenmark bereits messbar betroffen sind. In Deutschland und im übrigen Europa wird in der Praxis meist Binet verwendet, weil es schnell, übersichtlich und eng an der körperlichen Untersuchung bleibt.
Binet in der täglichen Praxis
Das Binet-System teilt die Erkrankung in drei Stadien ein. Entscheidend sind die Zahl der betroffenen Lymphknotenregionen sowie Anämie und Thrombozytopenie. Ich finde dieses System deshalb so nützlich, weil es mit wenigen Angaben bereits eine recht klare Orientierung gibt.
| Stadium | Kriterien | Praktische Einordnung |
|---|---|---|
| A | Hämoglobin ≥ 10 g/dl, Thrombozyten ≥ 100.000/µl, weniger als 3 betroffene Regionen | Frühe Erkrankung, oft ohne unmittelbaren Therapiebedarf |
| B | Hämoglobin ≥ 10 g/dl, Thrombozyten ≥ 100.000/µl, 3 oder mehr betroffene Regionen | Mehr Tumorlast, aber noch keine Knochenmarkinsuffizienz |
| C | Hämoglobin < 10 g/dl und/oder Thrombozyten < 100.000/µl | Fortgeschrittener Befall, häufig mit aktiver Erkrankung oder Knochenmarkversagen |
Mit den betroffenen Regionen sind typischerweise zervikale, axilläre und inguinale Lymphknoten sowie Milz und Leber gemeint. Wichtig ist mir ein Punkt besonders: Für die Einteilung zählt die körperliche Untersuchung und das Blutbild. Bildgebung kann im Einzelfall für Therapiefragen wichtig sein, sie verändert das Stadium selbst aber nicht. Genau das wird im Alltag oft verwechselt.
Lesen Sie auch: Melanom Stadium 1 - Prognose & Nachsorge: Was Sie wissen müssen
Rai als zweites Orientierungssystem
Das Rai-System arbeitet mit fünf Stadien und ist vor allem im US-amerikanischen Raum verbreitet. Inhaltlich beschreibt es dieselbe Idee etwas feiner: erst Lymphozytose, dann Lymphknoten- oder Organvergrößerung, später Zytopenien. Für die meisten Leser ist es sinnvoll, die Stadien eher als Übersetzungshilfe zu sehen als als getrennte Welten.
| Stadium | Kriterien | Praktische Einordnung |
|---|---|---|
| 0 | Nur Lymphozytose | Sehr frühe Erkrankung, meist Beobachtung |
| I | Lymphozytose plus Lymphadenopathie | Mehr Krankheitsausdehnung, aber noch keine Zytopenien |
| II | Lymphozytose plus Splenomegalie oder Hepatomegalie, mit oder ohne Lymphknotenbefall | Organbeteiligung ohne Knochenmarkversagen |
| III | Lymphozytose plus Anämie | Hinweis auf Knochenmarkbeteiligung oder relevante Blutbildungsstörung |
| IV | Lymphozytose plus Thrombozytopenie | Höheres Risiko für Beschwerden und Behandlungsbedarf |
Die eigentliche Kunst liegt danach nicht in der Zahl selbst, sondern in der richtigen Interpretation. Denn dieselbe Stadienzahl kann bei zwei Menschen sehr unterschiedlich aussehen, wenn Alter, Begleiterkrankungen und genetische Marker auseinandergehen. Genau dort wird die Prognose wirklich interessant.
Was die einzelnen Stadien für den Verlauf bedeuten
Die wichtigste Botschaft ist unspektakulär, aber zentral: Ein frühes Stadium bedeutet oft einen langsamen Verlauf, ein höheres Stadium bedeutet eher mehr Aufmerksamkeit, aber nicht automatisch einen dramatischen Verlauf. Ich würde Binet A oder Rai 0 nie mit „harmlos“ verwechseln, aber auch nicht mit „behandlungsbedürftig“. Bei CLL ist die Grenze zwischen Beobachten und Therapieren bewusst enger an Aktivität als an Stufennummern geknüpft.
| Einordnung | Typischer Verlauf | Was das praktisch heißt |
|---|---|---|
|
Frühes Stadium Binet A / Rai 0 |
Oft über längere Zeit stabil, manchmal jahrelang ohne Therapie | Regelmäßige Kontrollen sind wichtiger als sofortige Behandlung |
|
Intermediäre Stadien Binet B / Rai I-II |
Sehr variable Dynamik, von stabil bis klar progredient | Mehr Beobachtung, stärkere Aufmerksamkeit für Lymphknoten, Milz und Blutwerte |
|
Fortgeschrittene Stadien Binet C / Rai III-IV |
Häufig Zeichen von Knochenmarkversagen oder ausgeprägter Krankheitsaktivität | Therapie wird oft notwendig, die Ursache der Zytopenien muss genau geklärt werden |
Gerade bei Binet C und Rai III/IV lohnt die saubere Differenzierung: Ist die Anämie oder Thrombozytopenie durch eine direkte Knochenmarkinfiltration verursacht, oder steckt eine autoimmune Zytopenie dahinter? Beides ist klinisch relevant, aber prognostisch und therapeutisch nicht identisch. Diese Unterscheidung ist im Alltag oft wichtiger als die bloße Etikette „fortgeschritten“.
Ich formuliere es bewusst so klar: Stadium C ist nicht gleich hoffnungslos. Moderne zielgerichtete Therapien haben den Verlauf deutlich verändert, und selbst bei ungünstiger Ausgangslage kann die Erkrankung heute gut kontrollierbar sein. Die nächste Frage ist deshalb nicht nur, wie weit die CLL ist, sondern welche Biologie dahintersteht.
Warum die Prognose heute stärker von der Biologie als von der Zahl abhängt
Die Prognose bei CLL lässt sich nicht mehr seriös aus dem Stadium allein ableiten. Zwei Menschen mit demselben Binet-Stadium können völlig unterschiedliche Verläufe haben, wenn bei der einen Person ein mutierter IGHV-Status vorliegt und bei der anderen eine TP53-Aberration. Ich halte das für einen der wichtigsten Fortschritte der letzten Jahre: Wir sehen die Erkrankung feiner und können Risiken besser trennen.
| Prognostischer Faktor | Warum er wichtig ist | Was ich daraus ableite |
|---|---|---|
| TP53-Mutation oder del(17p) | Gehört zu den klar ungünstigen Merkmalen und ist mit kürzeren Remissionen verbunden | Therapiestrategie und Nachsorge müssen besonders sorgfältig gewählt werden |
| IGHV-mutiert vs. unmutiert | Unmutierter Status spricht meist für eine aktivere Biologie und kürzere krankheitsfreie Intervalle | Hilft, die Dynamik und Dauer des Ansprechens besser einzuordnen |
| Komplexer Karyotyp | Hinweis auf eine genetisch instabilere Erkrankung | Erhöht die Wachsamkeit, vor allem bei Therapieplanung und Rezidivrisiko |
| β2-Mikroglobulin | Spiegelt Tumorlast und biologische Aktivität wider | Nützlich als zusätzlicher Prognoseparameter |
| Alter und Komorbidität | Bestimmen, wie belastbar der Körper ist und welche Therapien sinnvoll sind | Die Prognose ist immer auch eine Frage der Gesamtgesundheit, nicht nur der Leukämie |
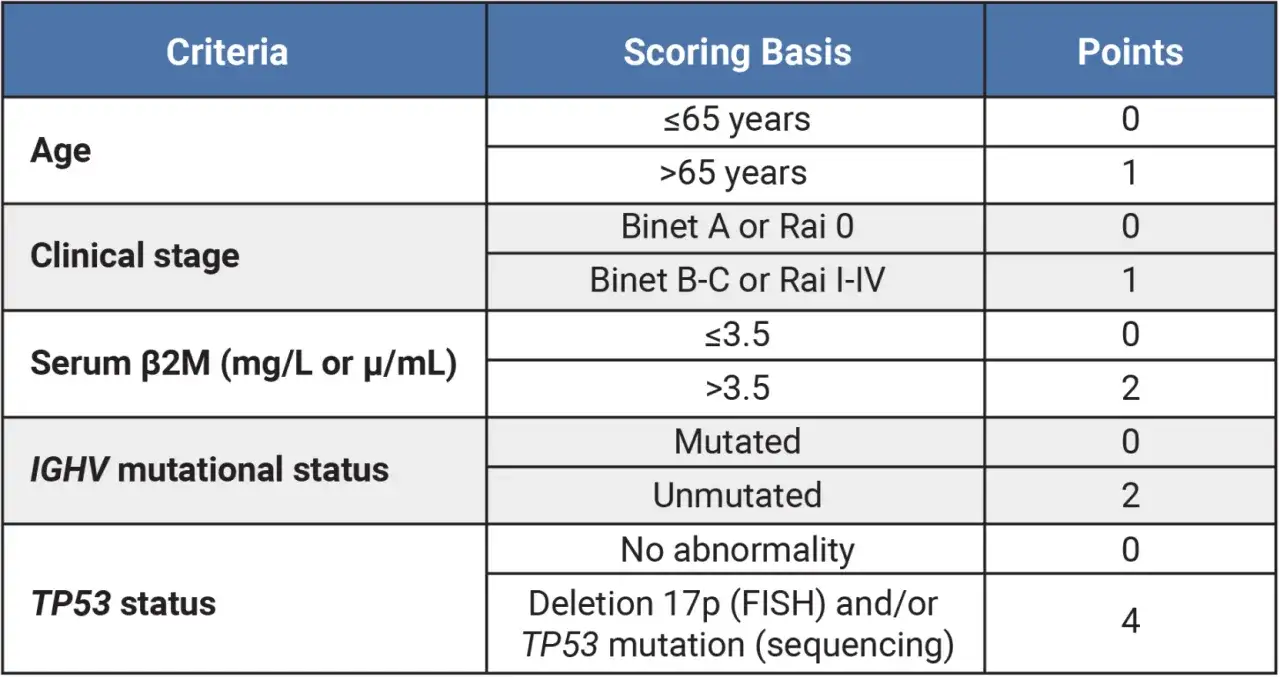
Vor einer Erstlinientherapie kann man mit dem CLL-IPI die Prognose grober abschätzen. Der Score berücksichtigt Alter, Stadium, β2-Mikroglobulin, IGHV und TP53/del(17p). Ich sehe ihn als hilfreiches Werkzeug für die Einordnung des progressionsfreien Überlebens, aber nicht als alleiniges Entscheidungssystem für die moderne Erstbehandlung. Dafür ist die aktuelle Therapielandschaft inzwischen zu differenziert. Gerade bei zielgerichteten Therapien bleibt die Prognose dadurch besser modellierbar, aber nie auf eine einzige Kennzahl reduzierbar.
Das ist auch der Grund, warum ich Prognosegespräche nie nur mit „gut“ oder „schlecht“ führe. Sinnvoller ist die Frage: Welche Faktoren sprechen für einen ruhigen Verlauf, welche für eine frühere Aktivität, und welche davon kann ich therapeutisch heute wirklich beeinflussen? Genau daran hängt die nächste Stufe der Entscheidung.
Wann Beobachtung reicht und wann Therapie sinnvoll wird
Bei asymptomatischer früher CLL ist Beobachten statt sofort behandeln Standard. Das ist kein Zögern und kein Versäumnis, sondern absichtlich so angelegt. Frühe antileukämische Therapie bringt ohne aktive Erkrankung keinen Überlebensvorteil, erhöht aber Nebenwirkungen und Bindung an die Behandlung. Ich halte es deshalb für einen Fehler, das Warten mit Untätigkeit zu verwechseln.
| Kriterium aktiver Erkrankung | Worauf es konkret ankommt |
|---|---|
| Knochenmarkversagen | Neu auftretende oder zunehmende Anämie und/oder Thrombozytopenie; Hb unter 10 g/dl oder Thrombozyten unter 100.000/µl sind meist ein klares Warnsignal |
| Milz oder Lymphknoten | Massive, progressive oder symptomatische Splenomegalie beziehungsweise Lymphadenopathie |
| Rasche Lymphozytose | Anstieg um mehr als 50 Prozent in zwei Monaten oder Lymphozytenverdopplungszeit unter 6 Monaten |
| Autoimmune Komplikationen | Autoimmunhämolyse oder Immunthrombozytopenie, wenn sie auf Kortison schlecht anspricht |
| Extranodaler Befall | Symptome an Haut, Niere, Lunge, Wirbelsäule oder anderen Organen |
| Allgemeinsymptome | Ungewollter Gewichtsverlust über 10 Prozent in 6 Monaten, Fieber über 38 °C über 2 Wochen, Nachtschweiß über 1 Monat, deutliche Fatigue |
Ein wichtiger Denkfehler ist, die absolute Lymphozytenzahl allein als Therapieauslöser zu sehen. Das ist sie nicht. Auch eine Hypogammaglobulinämie allein reicht nicht aus. Entscheidend ist das Gesamtbild aus Beschwerden, Organbefall, Blutbild und Dynamik. Wenn ich etwas im Verlauf besonders ernst nehme, dann nicht einen einzelnen Ausreißer, sondern die Kombination mehrerer Veränderungen.
Das gilt auch für den Rückfall: Nicht jeder erneute Anstieg der Lymphozyten verlangt sofort eine neue Behandlung. Maßgeblich bleibt, ob die Erkrankung wieder aktiv und klinisch relevant wird. Damit ist die Folgerung klar: Nicht der Laborwert allein steuert die Therapie, sondern die Frage, ob die CLL gerade wirklich die Biologie verändert.
Wie ich Verlauf und Prognose im Alltag einordnen würde
Für Betroffene ist die Diagnose meist dann am schwersten, wenn vieles noch unklar ist. In dieser Phase hilft mir eine nüchterne, aber vollständige Reihenfolge: Erst das Stadium verstehen, dann die Genetik einordnen, dann die Symptome im Alltag beobachten. Alles andere führt schnell zu Überinterpretationen einzelner Werte.
Bei den Kontrollen frage ich gedanklich immer dieselben Punkte ab: Ist das Stadium stabil? Gibt es Anzeichen für aktive Erkrankung? Haben sich Blutbild, Milz, Lymphknoten oder Allgemeinzustand verändert? Und liegen Risikomarker vor, die den Verlauf beschleunigen könnten? Besonders wichtig sind in meinen Augen diese Fragen:
- Welches Binet- oder Rai-Stadium liegt vor? Das ist die Basis, aber eben nicht die ganze Geschichte.
- Sind TP53/del(17p) und IGHV bestimmt? Ohne diese Werte bleibt die Prognose unvollständig.
- Wie hoch ist das β2-Mikroglobulin? Es ergänzt die Einordnung von Tumorlast und Risiko.
- Gibt es klinische Zeichen aktiver Erkrankung? Hier entscheidet sich, ob Beobachten noch ausreicht.
- Wie schnell verändern sich Blutbild und Beschwerden? Die Dynamik ist oft aussagekräftiger als ein Einzelwert.
Im Alltag halte ich außerdem nichts von übertriebener Bildgebung, wenn Klinik und Blutwerte ruhig sind. Die meisten Verläufe werden durch körperliche Untersuchung und Blutkontrollen erkannt, nicht durch dauernde CTs. Bildgebung bleibt sinnvoll, wenn etwas unklar ist oder ein spezieller Verdacht besteht, aber sie ersetzt kein gutes klinisches Follow-up. Gerade bei CLL ist das oft die ruhigere und zugleich präzisere Form der Überwachung.
Wer die Befunde mit diesen Fragen liest, erlebt die Erkrankung meist weniger diffus. Aus einem scheinbar abstrakten Stadium wird dann ein konkretes Bild: Was ist stabil, was ist biologisch auffällig, und was müsste sich ändern, damit sich der nächste Schritt ändert? Genau diese Klarheit macht in der Begleitung viel aus.
Welche Warnzeichen ich bei CLL nicht abwarten würde
Es gibt bei CLL Momente, in denen ich nicht auf den nächsten Routinetermin warten würde. Das heißt nicht automatisch, dass etwas Dramatisches passiert, aber solche Veränderungen gehören zeitnah abgeklärt. Besonders aufmerksam wäre ich bei folgenden Signalen:
- rasch größer werdende Lymphknoten oder ein deutlich zunehmender Druck im Bauch
- neues oder stärkeres Fieber, Nachtschweiß oder ungewollter Gewichtsverlust
- zunehmende Luftnot, starke Müdigkeit, Blässe oder Schwindel
- blaue Flecken, Nasenbluten oder punktförmige Hautblutungen
- wiederkehrende oder ungewöhnlich schwere Infekte
- plötzliche, klare Verschlechterung des Allgemeinzustands
Gerade eine schnelle Verschlechterung mit stark wachsenden Lymphknoten sollte immer an eine mögliche Transformation denken lassen und ärztlich bewertet werden. Unterm Strich gilt für mich: Die Stadieneinteilung gibt die Richtung vor, die Genetik schärft das Risiko, und die Symptome entscheiden oft über den richtigen Zeitpunkt für den nächsten Schritt. Wer diese drei Ebenen zusammen betrachtet, versteht den Verlauf der CLL deutlich realistischer als mit einer einzelnen Zahl im Befund.